The biopharmaceutical industry’s drug development success is impeded by a lack of physiological models that reflect the complexity of disease. HemoShear’s proprietary REVEAL-Tx™ platform combines biological and computational models of human disease to accelerate discovery of novel targets and successful new drug treatments.
Click below to learn more about HemoShear's platform.
HemoShear has built an extensive network for obtaining explanted healthy and diseased tissue under IRB-approved protocols for our drug discovery research. We partner with leading transplant centers within the organ donor system in real time to obtain tissue for targeted rare diseases. Our scientists isolate viable cells that are utilized for understanding and recapitulating diseases in our proprietary disease modeling system.
We leverage powerful computational techniques to incorporate data from our disease models and other sources to identify novel therapeutic targets. We then validate the computationally predicted targets in our disease models.
The HemoShear drug discovery team is comprised of more than 30 scientists. These experts apply their years of experience across biology, pharmacology, biomedical engineering, computational science, and medicinal chemistry to validate targets and characterize biological mechanisms to ultimately design and optimize drug candidates to move rapidly into the clinic.
We have developed translational models of human diseases by applying principles of physiological blood flow to human tissue obtained through our procurement network to recapitulate diseases for our programs. Our models enable valuable insights into complex disease pathways and allow for interrogation of drug responses in a more meaningful human-relevant context.

Propionic acidemia (PA) and methylmalonic acidemia (MMA) are rare autosomal recessive disorders of propionyl-CoA (P-CoA) catabolism, caused by a deficiency in the enzymes P-CoA carboxylase and methylmalonyl-CoA (M-CoA) mutase, respectively. PA and MMA are classified as intoxication-type inborn errors of metabolism because the intramitochondrial accumulation of P-CoA, M-CoA, and other metabolites results in secondary inhibition of multiple pathways of intermediary metabolism, leading to organ dysfunction and failure. Herein, we describe the structure–activity relationships of a series of short-chain carboxylic acids which reduce disease-related metabolites in PA and MMA primary hepatocyte disease models. These studies culminated in the identification of 2,2-dimethylbutanoic acid (10, HST5040) as a clinical candidate for the treatment of PA and MMA. Additionally, we describe the in vitro and in vivo absorption, distribution, metabolism, and excretion profile of HST5040, data from preclinical studies, and the synthesis of the sodium salt of HST5040 for clinical trials.
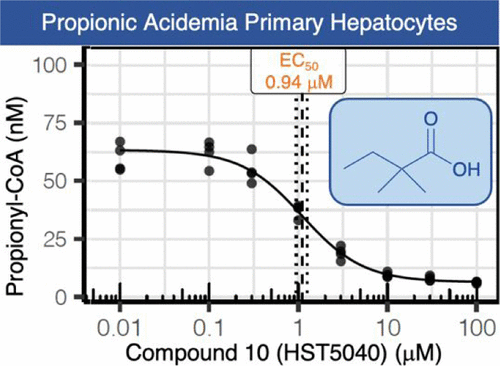

Propionic Acidemia (PA) and Methylmalonic Acidemia (MMA) are inborn errors of metabolism affecting the catabolism of valine, isoleucine, methionine, threonineand odd-chain fatty acids. These are multi-organ disorders caused by the enzymatic deficiency of propionyl-CoA carboxylase (PCC) or methylmalonyl-CoA mutase (MUT), resulting in the accumulation of propionyl-coenzyme A (P-CoA) and methylmalonyl-CoA (M-CoA in MMA only). Primary metabolites of these CoA esters include 2-methylcitric acid (MCA), propionyl-carnitine (C3), and 3-hydroxypropionic acid, which are detectable in both PA and MMA, and methylmalonic acid, which is detectable in MMA patients only (Chapman et al., 2012). We deployed liver cell-based models that utilized PA and MMA patient-derived primary hepatocytes to validate a small molecule therapy for PA and MMA patients. The small molecule, HST5040, resulted in a dose-dependent reduction in the levels of P-CoA, M-CoA (in MMA) and the disease-relevant biomarkers C3, MCA, and methylmalonic acid (in MMA). A putative working model of how HST5040 reduces the P-CoA and its derived metabolites involves the conversion of HST5040 to HST5040-CoA driving the redistribution of free and conjugated CoA pools, resulting in the differential reduction of the aberrantly high P-CoA and M-CoA. The reduction of P-CoA and M-CoA, either by slowing production (due to increased demands on the free CoA (CoASH) pool) or enhancing clearance (to replenish the CoASH pool), results in a net decrease in the CoA-derived metabolites (C3, MCA and MMA (MMA only)). A Phase 2 study in PA and MMA patients will be initiated in the United States.

Propionic acidemia (PA) and methylmalonic acidemia (MMA) are autosomal recessive disorders of propionyl-CoA (P-CoA) catabolism, which are caused by a deficiency in the enzyme propionyl-CoA carboxylase or the enzyme methylmalonyl-CoA (MM-CoA) mutase, respectively. The functional consequence of PA or MMA is the inability to catabolize P-CoA to MM-CoA or MM-CoA to succinyl-CoA, resulting in the accumulation of P-CoA and other metabolic intermediates, such as propionylcarnitine (C3), 3-hydroxypropionic acid, methylcitric acid (MCA), and methylmalonic acid (only in MMA). P-CoA and its metabolic intermediates, at high concentrations found in PA and MMA, inhibit enzymes in the first steps of the urea cycle as well as enzymes in the tricarboxylic acid (TCA) cycle, causing a reduction in mitochondrial energy production. We previously showed that metabolic defects of PA could be recapitulated using PA patient-derived primary hepatocytes in a novel organotypic system. Here, we sought to investigate whether treatment of normal human primary hepatocytes with propionate would recapitulate some of the biochemical features of PA and MMA in the same platform. We found that high levels of propionate resulted in high levels of intracellular P-CoA in normal hepatocytes. Analysis of TCA cycle intermediates by GC–MS/MS indicated that propionate may inhibit enzymes of the TCA cycle as shown in PA, but is also incorporated in the TCA cycle, which does not occur in PA. To better recapitulate the disease phenotype, we obtained hepatocytes derived from livers of PA and MMA patients. We characterized the PA and MMA donors by measuring key proximal biomarkers, including P-CoA, MM-CoA, as well as clinical biomarkers propionylcarnitine-to-acetylcarnitine ratios (C3/C2), MCA, and methylmalonic acid. Additionally, we used isotopically-labeled amino acids to investigate the contribution of relevant amino acids to production of P-CoA in models of metabolic stability or acute metabolic crisis. As observed clinically, we demonstrated that the isoleucine and valine catabolism pathways are the greatest sources of P-CoA in PA and MMA donor cells and that each donor showed differential sensitivity to isoleucine and valine. We also studied the effects of disodium citrate, an anaplerotic therapy, which resulted in a significant increase in the absolute concentration of TCA cycle intermediates, which is in agreement with the benefit observed clinically. Our human cell-based PA and MMA disease models can inform preclinical drug discovery and development where mouse models of these diseases are inaccurate, particularly in well-described species differences in branched-chain amino acid catabolism.

Background - Propionic acidemia (PA) is a disorder of intermediary metabolism with defects in the alpha or beta subunits of propionyl CoA carboxylase (PCCA and PCCB respectively) enzyme. We previously described a liver culture system that uses liver-derived hemodynamic blood flow and transport parameters to restore and maintain primary human hepatocyte biology and metabolism utilizing physiologically relevant milieu concentrations.
Methods - In this study, primary hepatocytes isolated from the explanted liver of an 8-year-old PA patient were cultured in the liver system for 10 days and evaluated for retention of differentiated polarized morphology. The expression of PCCA and PCCB was assessed at a gene and protein level relative to healthy donor controls. Ammonia and urea levels were measured in the presence and absence of amino acid supplements to assess the metabolic consequences of branched-chain amino acid metabolism in this disease.
Results - Primary hepatocytes from the PA patient maintained a differentiated polarized morphology (peripheral actin staining) over 10 days of culture in the system. We noted lower levels of PCCA and PCCB relative to normal healthy controls at the mRNA and protein level. Supplementation of branched-chain amino acids, isoleucine (5 mM) and valine (5 mM) in the medium, resulted in increased ammonia and decreased urea in the PA patient hepatocyte system, but no such response was seen in healthy hepatocytes or patient-derived fibroblasts.
Conclusions - We demonstrate for the first time the successful culture of PA patient-derived primary hepatocytes in a differentiated state, that stably retain the PCCA and PCCB enzyme defects at a gene and protein level. Phenotypic response of the system to an increased load of branched-chain amino acids, not possible with fibroblasts, underscores the utility of this system in the better understanding of the molecular pathophysiology of PA and examining the effectiveness of potential therapeutic agents in the most relevant tissue.

A barrier to drug development for nonalcoholic steatohepatitis (NASH) is the absence of translational preclinical human-relevant systems. An in vitro liver model was engineered to incorporate hepatic sinusoidal flow, transport, and lipotoxic stress risk factors (glucose, insulin, free fatty acids) with cocultured primary human hepatocytes, hepatic stellate cells (HSCs), and macrophages. Transcriptomic, lipidomic, and functional endpoints were evaluated and compared with clinical data from NASH patient biopsies. The lipotoxic milieu promoted hepatocyte lipid accumulation (4-fold increase, P< 0.01) and a lipidomics signature similar to NASH biopsies. Hepatocyte glucose output increased with decreased insulin sensitivity. These changes were accompanied by increased inflammatory analyte secretion (e.g., IL-6, IL-8, alanine aminotransferase). Fibrogenic activation markers increased with lipotoxic conditions, including secreted TGF-β (>5-fold increase, P < 0.05), extracellular matrix gene expression, and HSC activation. Significant pathway correlation existed between this in vitro model and human biopsies. Consistent with clinical trial data, 0.5 μM obeticholic acid in this model promoted a healthy lipidomic signature, reduced inflammatory and fibrotic secreted factors, but also increased ApoB secretion, suggesting a potential adverse effect on lipoprotein metabolism. Lipotoxic stress activates similar biological signatures observed in NASH patients in this system, which may be relevant for interrogating novel therapeutic approaches to treat NASH.

Lung cancer rates are rising globally and non-small cell lung cancer (NSCLC) has a five year survival rate of only 24%. Unfortunately, the development of drugs to treat cancer is severely hampered by the inefficiency of translating pre-clinical studies into clinical benefit. Thus, we sought to apply a tumor microenvironment system (TMES) to NSCLC. Using microvascular endothelial cells, lung cancer derived fibroblasts, and NSCLC tumor cells in the presence of in vivo tumor-derived hemodynamic flow and transport, we demonstrate that the TMES generates an in-vivo like biological state and predicts drug response to EGFR inhibitors. Transcriptomic and proteomic profiling indicate that the TMES recapitulates the in vivo and patient molecular biological state providing a mechanistic rationale for the predictive nature of the TMES. This work further validates the TMES for modeling patient tumor biology and drug response indicating utility of the TMES as a predictive tool for drug discovery and development and potential for use as a system for patient avatars.
HemoShear has more than 30 publications validating our platform in a range of scientific applications. For more information, contact info@hemoshear.com.
HemoShear Therapeutics, Inc.
501 Locust Avenue
Suite 301
Charlottesville, Virginia 22902